
The Covid-19 Event brought several new terms into common currency:
Social Distancing
Contact Tracing
Lockdown
Coronavirus
Flatten the Curve
Shelter in Place
PCR is another term which most people had never heard of before 2020 but which has now entered the public consciousness. You may even know that the letters stand for Polymerase Chain Reaction and that this is a test used to confirm whether or not a patient has COVID-19. For most of 2020, PCR was the test for COVID-19—the “gold standard”, as it were. It was quite late in the pandemic that other kinds of tests—antigen tests and antibody tests, for example—became widely available.
What, then, is PCR and is it as reliable as the public has been led to believe? That’s what we will be looking at in the next few articles.

Kary Mullis
The polymerase chain reaction was developed in 1983 by the American biochemist Kary Mullis, earning him a Nobel Prize in Chemistry in 1993. In his colourful autobiography, Dancing Naked in the Mind Field, Mullis describes how he first conceived of the PCR. At the time, he was working for Cetus Corporation, a biotech company in Emeryville, California. He was in charge of the synthesis of short chains of DNA, or oligonucleotides, for research purposes. His problem was to find a way of using oligonucleotides to synthesize a longer sequence of DNA—such as an entire gene, which might be hundreds of times as long as the oligonucleotides.
The Eureka moment came to him as he was driving along California’s Highway 128 one balmy summer night in 1983, with his girlfriend Jennifer Barnett dozing in the seat beside him:
The key to the problem lies in the oligonucleotides that my laboratory at Cetus now easily makes. Like a “FIND” sequence in a computer search, a short string of nucleotides in a synthetic molecule might be able to define a position along a very much longer natural DNA molecule. Finding a place to start is of utmost importance. Natural DNA is a tractless coil, like an unwound and tangled audio tape on the floor of the car in the dark.
What kind of chemical program would be required to “FIND” a specific sequence on DNA with 3 billion nucleotides and then display that sequence to a human who was trillions of times larger than the DNA? Instead of a list of statements in BASIC or FORTRAN run on a computer and displayed on a screen, I had to arrange a series of chemical reactions, the result of which would represent and display the sequence of a stretch of DNA. The odds were long. Like reading a particular license plate out on Interstate 5 at night from the moon.

I knew computer programming, and from that I understood the power of a reiterative mathematical procedure. That’s where you apply some process to a starting number to obtain a new number, and then you apply the same process to the new number, and so on. If the process is multiplication by two, then the result of many cycles is an exponential increase in the value of the original number: 2 becomes 4 becomes 8 becomes 16 becomes 32 and so on.
If I could arrange for a short synthetic piece of DNA to find a particular sequence and then start a process whereby that sequence would reproduce itself over and over, then I would be close to solving my problem.
The concept was not out of the question because in fact one of the natural functions of DNA molecules is to reproduce themselves. They do it every time a cell divides into two daughter cells. A short piece of synthetic DNA could be treated in such a way that it would stick to a longer strand of DNA in a specific way if the sequences matched up somewhere on the long piece. The matching process would not be perfect. I might locate a thousand different places that were similar to the one I was searching for in addition to the correct one. A thousand out of the 3 billion in the human genome would be no trivial feat, but it wouldn’t be enough. I needed to find just one place.
Suddenly, I knew how to do it. If I could locate a thousand sequences out of billions with one short piece of DNA, I could use another short piece to narrow the search. This one would be designed to bind to a sequence just down the chain from the first sequence I had found. It would scan over the thousand possibilities out of the first search to find just the one I wanted. And using the natural properties of DNA to replicate itself under certain conditions that I could provide, I could make that sequence of DNA between the sites where the two short search strings landed reproduce the hell out of itself. In one replicative cycle I could have two copies, and in two cycles I could have four, and in ten cycles ... I thought I remembered that two to the tenth was around a thousand ...
We were at mile marker 46.58 on Highway 128, and we were at the very edge of the dawn of the age of PCR ...

About a mile down the canyon, I pulled off again. The thing had just exploded again. A new and wonderful possibility. Not only could I make a zillion copies, but they would always be the same size. That was important. That was the almighty, the halleluja! clincher. The hell with Jennifer. I had just solved the two major problems in DNA chemistry. Abundance and distinction. And I had done it in one stroke. I stopped the car at a nice comfortable turnout and took my time working my way through the consequences. This simple technique would make as many copies as I wanted of any DNA sequence I chose, and everybody on Earth who cared about DNA would want to use it. It would spread into every biology lab in the world.
I would be famous. I would get the Nobel Prize. (Mullis 5-7)
The laboratory technique that Mullis had just come up with was primarily a way of synthesizing relatively short chains of nucleotides—the building blocks of DNA—such as a single gene, which might comprise a few thousand nucleotides. This, after all, is what Mullis did at Cetus. But as he makes clear in his account, PCR also resembles the “FIND” process one uses when one is searching through a computer file for a specific word or string of characters. Therefore, to paraphrase Mullis, it is not out of the question that this technique could be used to search a sample for the presence of a short but specific sequence of nucleotides—for example, a gene known to be unique to a particular pathogen. In other words, PCR could conceivably be used as a diagnostic tool to confirm the presence of a specific pathogen in a patient presenting in the ER with symptoms of uncertain etiology.
In fact, the purpose for which PCR was invented might not be the ideal use to which it could be put:
... it had not been my intention to revolutionize the world of biochemistry when I invented PCR; PCR was a tool I created because I needed to do an experiment. In truth, I was terribly naïve ... and if I had had more knowledge about what I was doing, PCR would never have been invented. (Mullis 24)
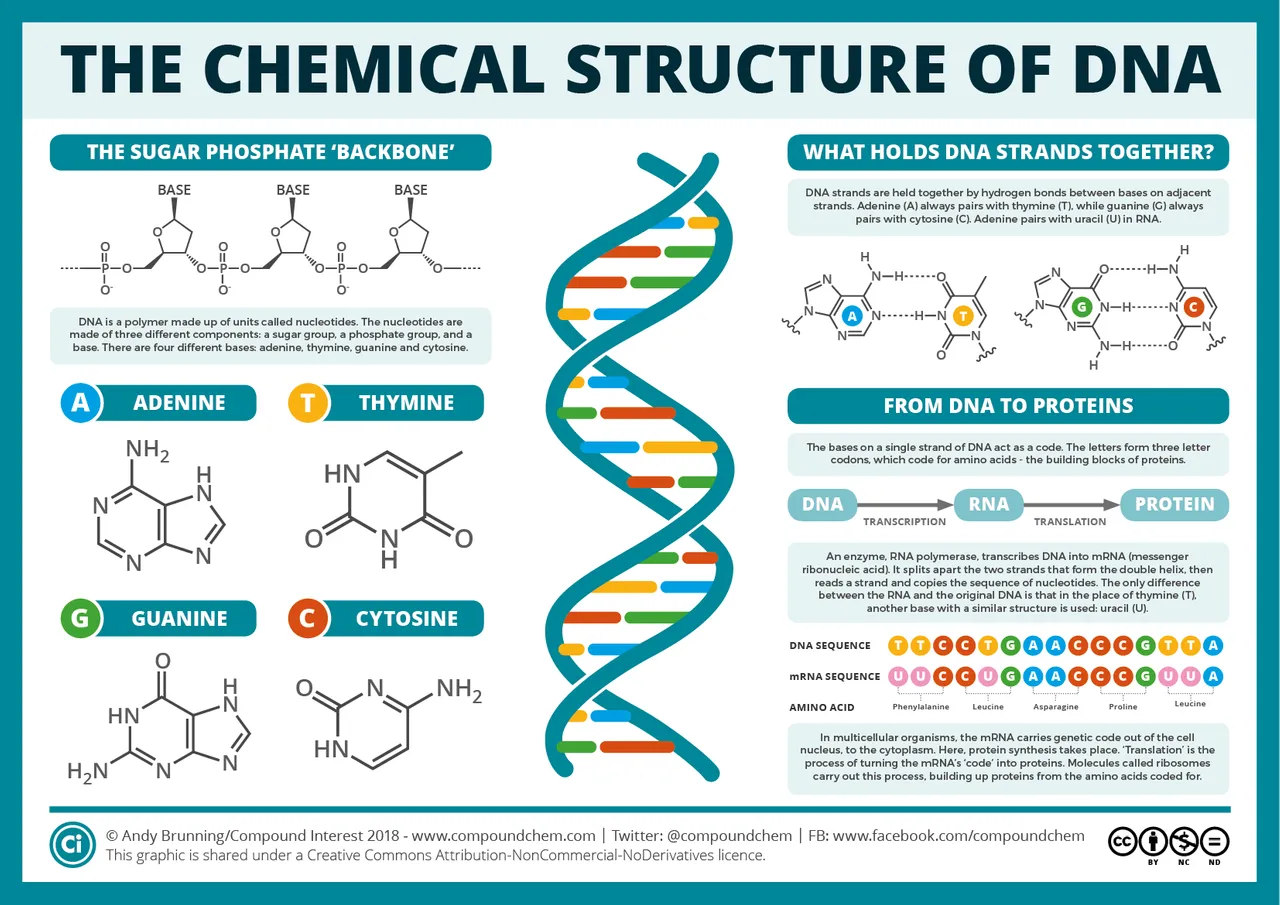
DNA
DNA, or deoxyribose nucleic acid, was discovered by the Swiss physician Friedrich Miescher in 1869. Because it is found in the nuclei of biological cells, it is called nucleic, giving DNA its N. The DNA molecule is a double helix—a twisted ladder—made of two complementary strands of nucleotides. Each nucleotide is comprised of three subunits:
A nucleobase. There are four different nucleobases: adenine, thymine, cytosine and guanine.
A pentose, or sugar containing a ring of five carbon atoms. In DNA this pentose is always deoxyribose, giving DNA its D.
A phosphate group. This consists of one, two or three phosphate ions—PO4. These are responsible for the acidic nature of DNA, giving DNA its A.
The two strands of the double helix are complementary and run in opposite directions. By convention, the upstream end of a strand of DNA is called the 5' end (“five prime end”). The downstream end is called the 3' end (“three prime end”). These numbers refer to the fact that the first carbon atom at the 5' end is the fifth carbon in the sugar-ring of deoxyribose, while the first carbon atom at the 3' end is the third carbon in the sugar-ring.
When two strands of DNA bind (anneal is the technical term) to form a double helix, the rungs of the ladder are made of base pairs—two nucleobases bound to each other by hydrogen bonds—while the pentose and phosphate groups form the side rails, or backbone, of the molecule. In DNA, there are two kinds of base pair: adenine-thymine and cytosine-guanine. Adenine always binds to thymine, and vice versa. Cytosine always binds to guanine, and vice versa. Because each nucleobase binds to one and only one other kind of nucleobase, the two strands of DNA are complementary: the sequence of bases on one strand of the double helix determines the sequence of bases on the complementary strand.
The two complementary strands of a double helix are referred to as the positive-sense and negative-sense strands. (The terms sense and antisense are also found in the literature.) Which is which depends on the way in which the DNA is replicated during cell division.
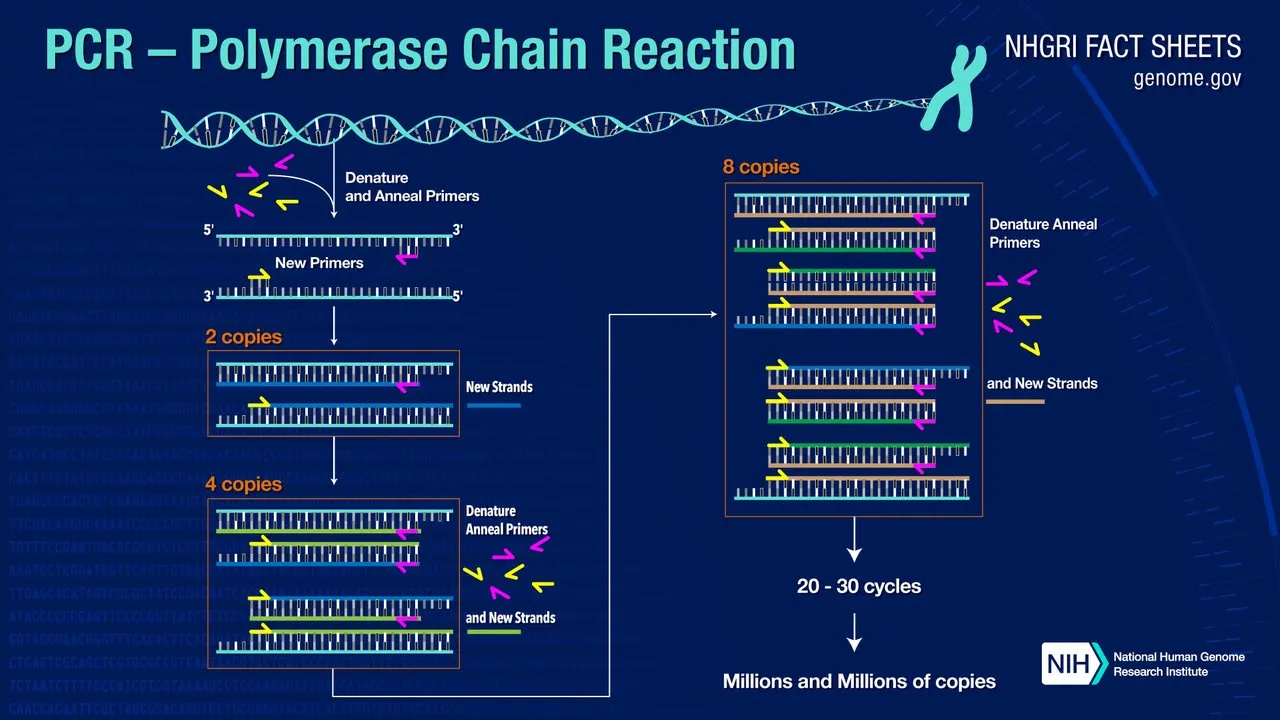
The Polymerase Chain Reaction
PCR is a chemical process that replicates a specific segment of DNA—the target sequence—producing millions or billions of copies of this sequence. Typically, the target sequence comprises 100-10,000 nucleotides (or base-pairs). The DNA template that contains the target sequence may comprise millions of sequences. It is a single DNA molecule, such as a chromosome. Mullis was primarily interested in quickly amplifying a very small sample of DNA—a single gene, for example—to such an extent that it could be studied in detail and used in experiments.
Most methods of PCR rely on thermal cycling, in which reactants undergo repeated cycles of heating and cooling. This permits different temperature-dependent reactions to take place—specifically, DNA melting or denaturation and enzyme-driven DNA replication or polymerization. PCR employs two main reagents: a pair of primers and a DNA polymerase.
Primers are relatively short single-strand fragments of DNA (or RNA, as we shall see in a later article). Each primer forms a complementary sequence to a piece of the template. Primers typically comprise only 18-24 nucleotides. One primer—the forward primer—binds to the 3' end of the target sequence on the negative-sense strand of DNA. The other primer—the reverse primer—binds to the 3' end of the complementary target sequence on the positive-sense strand. As we have seen, the target sequence itself may comprise hundreds or even thousands of nucleotides.
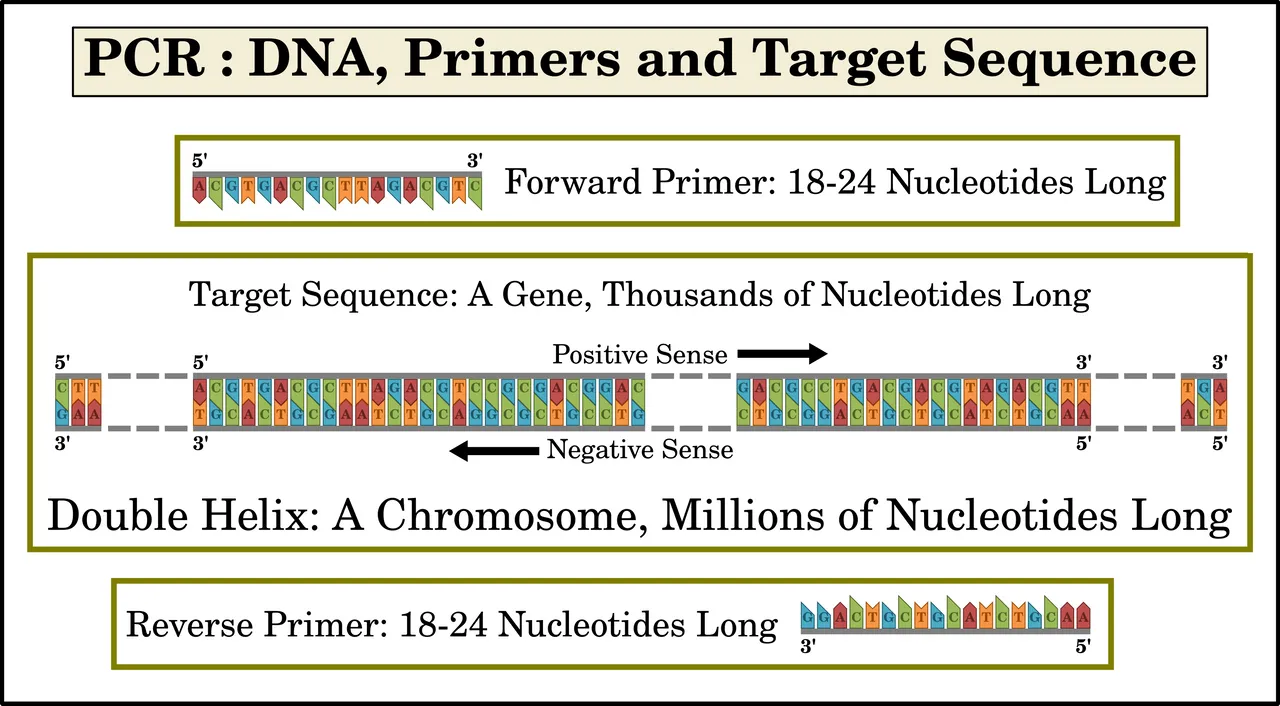
Taq polymerase is a heat-resistant enzyme that catalyzes the creation of new DNA strands by replicating—polymerizing—the target sequence. It was named after the heat-tolerant bacterium from which it was first isolated, Thermus aquaticus. It works by extending a short strand of DNA which is bound to a longer strand, using the longer strand as a template. In PCR, the primers bind to the long strands of DNA template. The primers are then extended along the template. Mullis’s great insight was his realization that a combination of two primers—one for the positive-sense target sequence and the other for the complementary negative-sense sequence—would result in the exponential replication of only the desired target sequence. The details of why this is so will be explained below and in the next article.
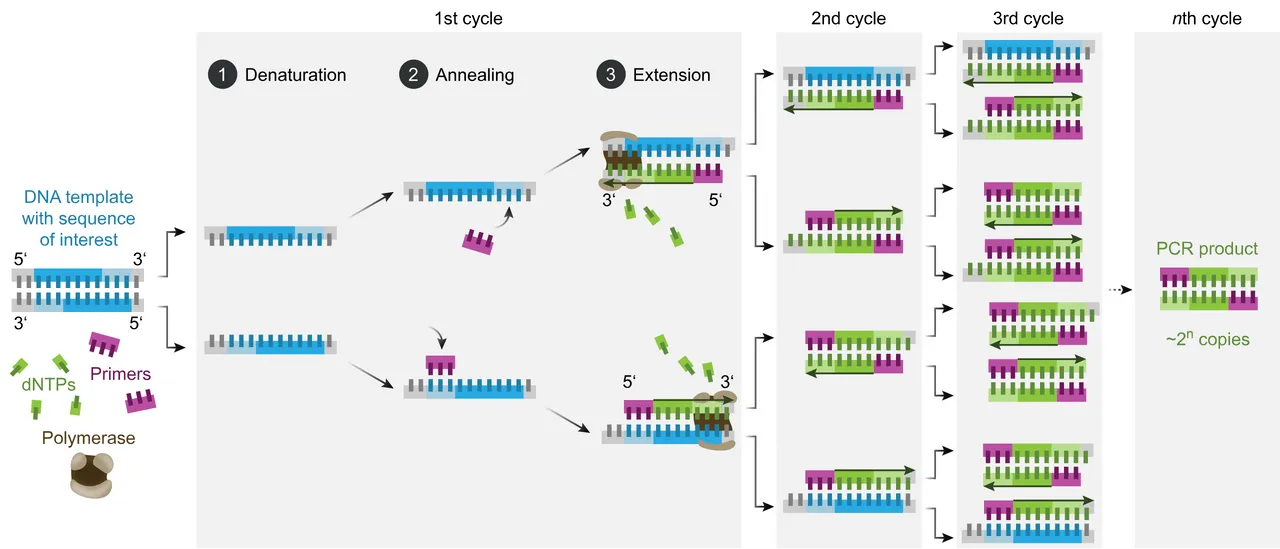
There are three steps in each cycle of the polymerase chain reaction:
Step 1: Denaturation In the first step, the two strands of the DNA double helix are physically separated at a high temperature (~ 95° C) in a process called nucleic acid denaturation or melting. This breaks the hydrogen bonds between the base pairs, creating long single strands of DNA. In subsequent cycles, any strands of double helix created in earlier cycles are also separated into single strands.
Step 2: Annealing In the second step, the temperature is lowered (~ 48-65° C), allowing the primers to bind to the strands of DNA at the places where the primer sequences complement the DNA sequences. The actual temperature will depend on the specific primers used. At the start of the reaction, there are typically millions of each primer for every strand of the DNA template. This is necessary because a pool of free-floating primers will be required in each cycle of the PCR. And the number of primers required will be doubling with each cycle. The huge excess of primers over DNA templates also means that there will be little renaturation of the original DNA strands. That is, the original double helices won’t reform, cancelling out the denaturation step. Some DNA renaturation may occur, but the huge excess of primers will enable them to outcompete the original strands of DNA.
Step 3: Extension In the final step, the temperature is raised again (~ 72° C) so that the Taq polymerase enzyme can catalyze the replication of the DNA. The replicated fragments are assembled from a pool of free-floating nucleotides that are always found in the presence of DNA. These free nucleotides are called deoxynucleoside triphosphates, or dNTPs, because they contain three phosphate groups.
When the cycle is repeated, the replicated fragments of DNA become templates for further replication, setting in motion a chain reaction in which the original target sequence is exponentially amplified. In general, each cycle doubles the number of replicated fragments, but the chain reaction will ultimately be limited by the number of free nucleotides available for assembly.
Extension
Extension is so called because replication extends the primers along the strand of DNA in the 5' to 3' direction, turning each primer into a longer strand of DNA. In other words, the 3' end of the primer is extended. If this process is allowed to run indefinitely, it will extend all the way to the end of the strand of DNA template, well beyond the end of the target sequence. If, however, it is only allowed to run for a limited time—determined by the length of the target sequence—it will be interrupted before it extends too far beyond the end of the target sequence. Typically, Taq polymerase replicates a thousand bases per minute. Nevertheless, after the first cycle is complete, the replicated strands will have the target sequence at their 5' ends, but their 3' ends will extend well beyond the far end of the target sequence.
The temperature is then raised to initiate the denaturation step of the next cycle. This results in single strands of DNA again. Half of these are the original long templates: sequences of millions of nucleotides, with the short target sequence somewhere in the middle. The other half are the replicated strands. These include shorter sequences containing the target sequence at their 5' ends, but whose 3' ends extend well beyond the far end of the target sequence. Some replicated strands will not contain the target sequence, but will contain a sequence of 18-24 nucleotides that complement one of the primers.
When the annealing step of the second cycle occurs, forward primers will anneal to reverse primers that were extended in the previous cycle—and vice versa. These forward primers, when extended in the second cycle, will only reach the far end of the target sequence, where the extension step of cycle one began. Similarly, when the reverse primers are extended in the second cycle, the extension will end when it reaches the far end of the target sequence, where the extension step of cycle one began. In both cases, the resulting strand will comprise only the target sequence. This is what made Mullis’s discovery worthy of a Nobel Prize. He had found a way to replicate easily and quickly a single gene thousands of nucleotides long using a couple of primers that were only 18-24 nucleotides long. By “bookending” the target sequence with two different primers, Mullis could ensure that any other section of DNA that contained a sequence of nucleotides complementary to only one of the primers would not be replicated.
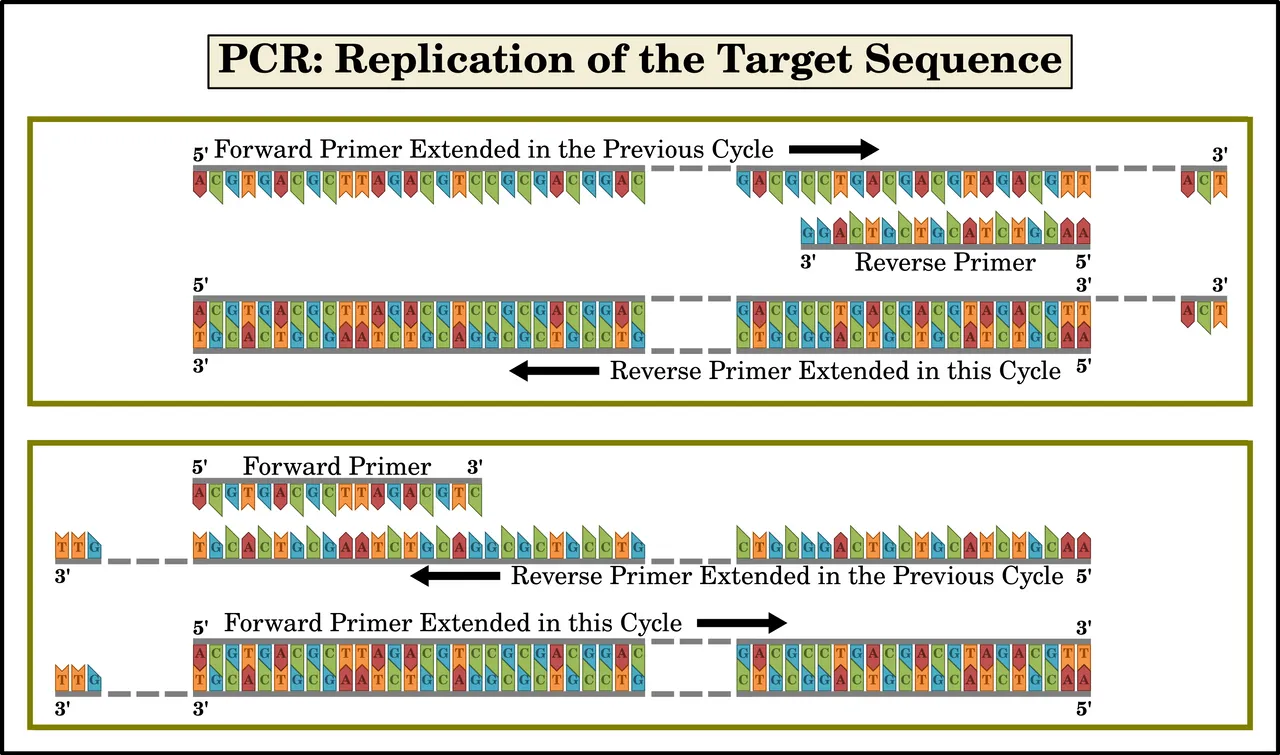
Of course, in each cycle, some primers will anneal to the original strands of the DNA template. When extended, these will result in replicated fragments much longer than the target sequence, just as in cycle one. So, each time a cycle is performed:
Very long strands of the original DNA template give rise to longish strands that include the target sequence at one end but whose other end goes well beyond the end of the target sequence. They may also give rise to longish strands which do not contain the target sequence but which do contain a sequence that complements only one of the primers.
Longish strands that include the target sequence at one end but whose other end goes well beyond the target sequence give rise to short strands that only contain the target sequence.
Short strands that only contain the target sequence give rise to new short strands that only contain the target sequence.
Longish strands which do not contain the target sequence but which do contain a sequence that complements one of the primers will not be annealed by the other primer, so these will not be replicated in this or any subsequent cycle.

The result is that each time a new cycle is performed, a greater proportion of the replicated strands comprise only the target sequence. The number of very long strands—the original DNA template—never increases. The number of longish strands increases, but only arithmetically (2, 4, 6, 8, 10, 12, ...). The number of short strands—the target sequence—increases geometrically (2, 4, 8, 16, 32, 64, ...).
Note that the exponential replication of the target sequence will take place if and only if a forward primer finds a complementary sequence to bind to on an extended reverse primer, and vice versa. If there are no complementary sequences for the primers to bind to, no replication will take place and there will be no exponential replication of the target sequence no matter how many times the PCR is cycled. This is why Mullis initially thought of PCR as a sort of FIND function: if the target sequence is present to begin with, it will be amplified exponentially : if it is not amplified exponentially, it was not present to begin with. This is the basis of the PCR test.
Uniqueness of Primers
In his account of how he first conceived of PCR, Kary Mullis writes the following about what we now call primers:
A short piece of synthetic DNA could be treated in such a way that it would stick to a longer strand of DNA in a specific way if the sequences matched up somewhere on the long piece. The matching process would not be perfect. I might locate a thousand different places that were similar to the one I was searching for in addition to the correct one. A thousand out of the 3 billion in the human genome would be no trivial feat, but it wouldn’t be enough. I needed to find just one place.
Suddenly, I knew how to do it. If I could locate a thousand sequences out of billions with one short piece of DNA, I could use another short piece to narrow the search. This one would be designed to bind to a sequence just down the chain from the first sequence I had found. It would scan over the thousand possibilities out of the first search to find just the one I wanted. And using the natural properties of DNA to replicate itself under certain conditions that I could provide, I could make that sequence of DNA between the sites where the two short search strings landed reproduce the hell out of itself. (Mullis 6)

Here, Mullis is talking about the forward primer (A short piece of synthetic DNA) and the reverse primer (another short piece ... designed to bind to a sequence just down the chain from the first sequence). He then goes on to say: I could make that sequence of DNA between the sites where the two short search strings landed reproduce the hell out of itself. Here, he is referring to the target sequence, which does indeed lie between the forward and reverse primers. And it is the target sequence that is exponentially replicated.
The earlier statement, then—I might locate a thousand different places that were similar to the one I was searching for in addition to the correct one—refers to the fact that because the primers are so short (only 18-24 nucleotides long), they are bound to find hundreds of places along the huge denatured strands of DNA (millions of nucleotides long) to bind to during the annealing step of cycle one. But when the extension step of cycle one has taken place, strands created by the extension of forward primers become templates for reverse primers in the next cycle—and vice versa. So unless the reverse primer can find the correct sequence along that strand, it will not bind to it and no extension will take place.
In other words, the trick of using two short primers, one of which is corresponds to the beginning (upstream or 5' end) of the target sequence on the positive sense strand and the other of which corresponds not to the end of the target sequence but to the beginning (upstream or 5' end) of the target sequence on the negative sense strand, ensures that after many cycles have been completed only the target sequence will be exponentially replicated—assuming, that is, that it was present somewhere along the original DNA template to begin with.
And that’s a good place to stop.
References
- Kary Banks Mullis, Nobel Lecture: The Polymerase Chain Reaction, (1993)
- Kary Banks Mullis, Dancing Naked in the Mind Field, Pantheon Books, New York (1998)
Image Credits
- COVID-19 Poster: © 2021 Dublin Region Homeless Executive, Fair Use
- Kary Mullis: © Dona Mapston (photographer), Creative Commons License
- California Highway 128, Between Cloverdale and Boonville: © 2022 Google, Fair Use
- California Highway 128, Mile Marker 45.64: © 2022 Google, Fair Use
- The Structure of DNA: © Andy Brunning/Compound Interest 2018, Creative Commons License
- The Polymerase Chain Reaction: © National Human Genome Research Institute, Fair Use
- Diagrammatic Representation of the Forward and Reverse Primers: Adapted from File:Primers RevComp.svg, © Richard Wheeler, Creative Commons License
- Target Sequence Replication: Adapted from File:Primers RevComp.svg, © Richard Wheeler, Creative Commons License
- The PCR Cycle: © Enzoklop (creator), Creative Commons License
- :
- Kary Mullis Receives the Nobel Prize from King Carl XVI Gustaf in Stockholm: © Bridgeman Images, Fair Use
Online Resources
- Kary Mullis on PCR: Timestamp = 48:37
- Kary Banks Mullis
- How Does Taq Polymerase Know When to Stop?
